
According to the Centers for Disease Control (CDC), sickle cell disease (SCD) affects millions of people throughout the world. It is particularly common among those whose ancestors came from sub-Saharan Africa, Spanish-speaking regions in the Western Hemisphere (South America, the Caribbean, and Central America), Saudi Arabia, India, and Mediterranean countries such as Turkey, Greece, and Italy. [i]
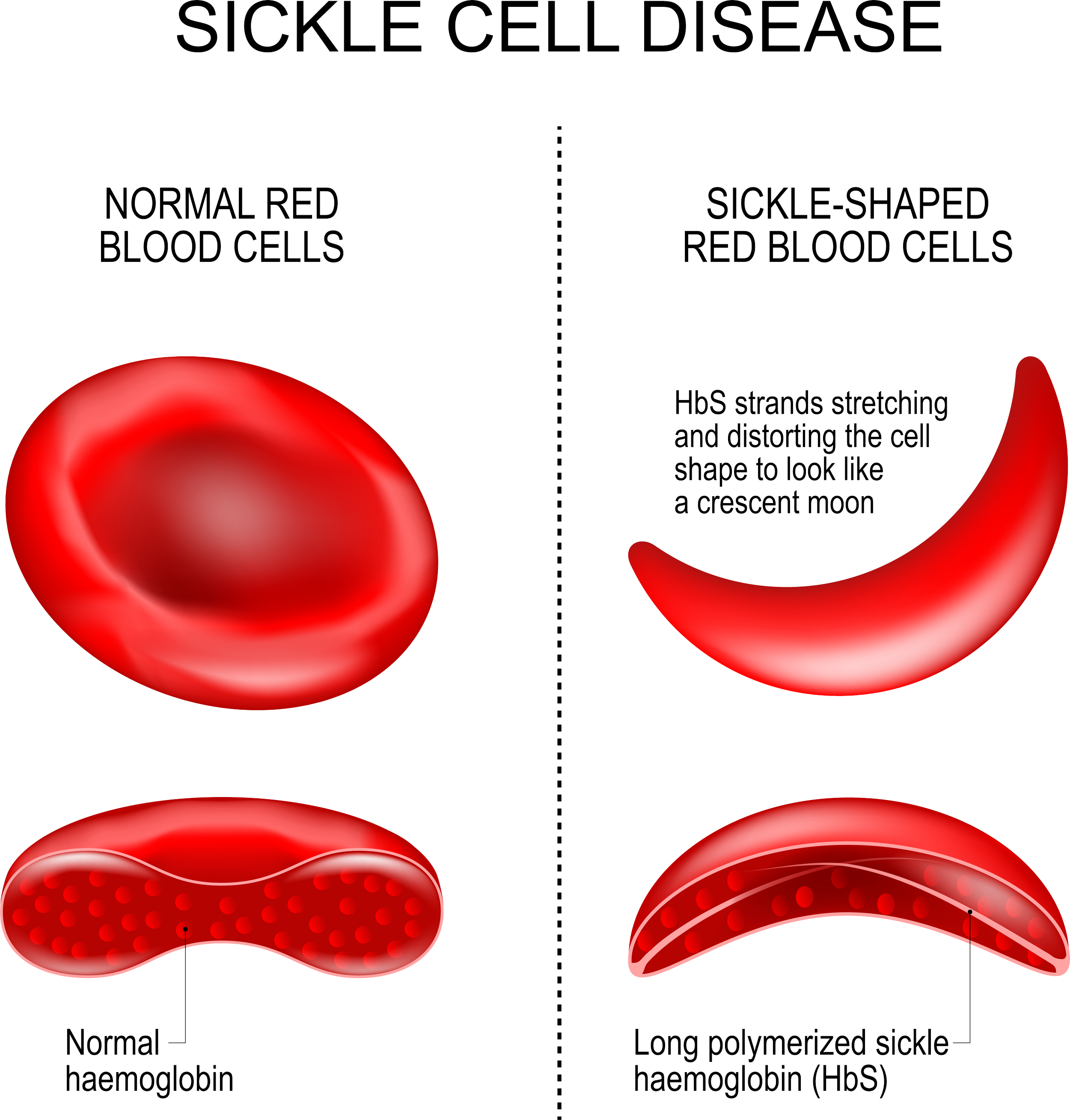
Every June 19, we observe World Sickle Cell Day to raise international awareness of sickle cell disease and the challenges patients and families face when handling the disease. Sickle cell disease is a group of inherited red blood cell (RBC) disorders that affect hemoglobin, the protein that carries oxygen through the body, and is caused by a genetic mutation of the HBB gene responsible for the β-globin component of hemoglobin found on chromosome 11. In SCD, the red blood cells to become hard and sticky and look like a sickle. The sickle cells die early, which causes a constant shortage of red blood cells. As the RBCs travel through small blood vessels, they get stuck and obstruct the blood flow. Sickle cell disease affects about 100,000 Americans, and is the most common inherited blood disorder in the U.S. According to CDC statistics[ii]
SCD occurs in about 1 out of every 365 African-American births
SCD occurs in about 1 out of every 16,300 Hispanic-American births
About 1 in 13 African-American babies is born with sickle cell trait (SCT)
There are three main types of SCD[iii] :
HbSS- inherits one gene, “S”, from each parent. This is the most common and most severe form.
HbSC- inherits “S” gene from one parent, and another gene “C” for a different type of abnormal hemoglobin from the other parent. This is a milder form of SCD.
HbS beta thalassemia- inherits “S” gene from one parent, and a gene for beta thalassemia from the other parent. Within this type of SCD, there are an additional two types: “0” which is the more severe form, and “+” which is a milder form of SCD.
SCD can cause acute chest syndrome, anemia, avascular necrosis, blood clots, dactylitis, fever, infection, kidney problems, leg ulcers, liver problems, organ damage, pain, priapism, pulmonary hypertension, sleep-disordered breathing, splenic sequestration, stroke, and vision loss. On average, it may lower life expectancy by 20-30 years.[iv] Early diagnosis and treatment is vital for SCD; therefore, newborn screening is mandated in all U.S. states.
Early diagnosis, preventative care, and effective approaches to treatment of SCD related complications have increased the average life span from 14 years old in the early 1970s, to well into the 40s.[v] The only cure at this time is a matched related donor stem cell transplant; unfortunately, only a small number of patients with SCD have a relative that is well-matched, which is required for the best chance of a successful transplant.[vi]
Currently there are two gene therapies awaiting biological license application (BLA) acceptance from the Food and Drug Administration (FDA): lovotibeglogene autotemcel (Lovo-cel) and exagamglogene autotemcel (Exa-cel). If the FDA accepts these BLA submissions, a decision could be made within eight months and the therapies available in 2024. Both therapies require mobilization and collection of stem cells, and then myeloablative conditioning with busulfan prior to infusion of the gene therapy.
Bluebird submitted its BLA for Lovo-cel on April 23, 2023. The BLA for Lovo-cel targets SCD patients 12 years and older with a history of vaso-occlusive events. Lovo-cel is designed to add functional copies of a modified form of the β-globin gene (βA-T87Q) into a patient’s own hematopoietic stem cells. It is comprised of autologous transplantation of hematopoietic stem and progenitor cells transduced with the BB305 lentiviral vector encoding a modified β‐globin gene to produce anti‐sickling hemoglobin (HbAT87Q).The goal is reducing sickled RBCs, hemolysis, and other complications.[vii] Per Bluebird’s website, the BLA submission is based on efficacy results from 36 patients in the HGB-206 Group C cohort with a median 32 months of follow-up and two patients in the HGB-210 study with 18 months of follow-up each. The BLA submission also includes safety data from 50 patients treated across the entire lovo-cel program, including six patients with six or more years of follow-up.[viii]

Source: SparkSickleCellChange.com
Vertex Pharmaceuticals and CRISPR Therapeutics completed their rolling BLA submission for Exa-cel on April 3, 2023, and are awaiting BLA acceptance from the FDA. The BLA for Exa-cel targets patients ages 12-35 with SCD, in addition to patients with transfusion-dependent beta thalassemia (TDT). Exa-cel is the first BLA submission to use a gene-editing technology, known as CRISPR/Cas9, which edits a person’s hematopoietic stem cells to produce fetal hemoglobin (HbF; hemoglobin F), thus increasing the amount of healthy hemoglobin in red blood cells. Per Vertex Pharmaceutical’s website, the aim is to use the body’s own machinery to switch back to producing fetal hemoglobin.[ix,x] The BLA submission efficacy was based on the open-label Phase 2/3 CLIMB–121 study which enrolled patients, ages 12–35, with severe SCD, defined as having at least two severe vaso-occlusive crises (VOCs) per year in the two years before enrollment. All participants received a single dose of exa-cel and are being followed for about two years. Results showed that a single dose of exa-cel increased fetal hemoglobin levels and prevented VOCs in these patients. Another pair of open-label Phase 3 trials is currently evaluating the treatment’s safety and effectiveness in pediatric patients, ages 5–11, with plans to eventually expand to patients as young as 2. CLIMB–151 (NCT05329649) is recruiting up to 12 children with SCD at sites in the U.K., U.S., and Italy.[xi]

Source: SparkSickleCellChange.com
In the 113 years since the discovery of SCD, progress has been slow in developing effective treatments for this debilitating disease. Due to the limited availability of matched related donors, stem cell transplant has not been an option for many patients. Recently, major advancements in technology have opened the door to gene therapy, bringing with it hope for a future cure.
Article by Kathy Clark, RN, BSN, CMCN, Vice President, Director of Managed Care. For more information about how this may affect your plan, please contact your Summit ReSources care specialist. The following sources were used as reference material for this article:
i CDC, Data & Statistics on Sickle Cell Disease, Accessed 6/5/2023, https://www.cdc.gov/ncbddd/sicklecell/data.html
ii CDC, Data & Statistics on Sickle Cell Disease, Accessed 6/5/2023, https://www.cdc.gov/ncbddd/sicklecell/data.html
iii CDC, What is Sickle Cell Disease, Accessed 6/5/2023, https://www.cdc.gov/ncbddd/sicklecell/facts.html
iv CDC, Complication of Sickle Cell Disease, Accessed 6/5/2023, https://www.cdc.gov/ncbddd/sicklecell/complications.html
v National Heart, Lung, and Blood Institute, A Century of Progress, Accessed 6/5/2023, https://www.nhlbi.nih.gov/resources/sickle-cell-disease-milestones-research-and-clinical-progress
vi National Institute of Health, Sickle Cell Disease, Accessed 6/5/2023, https://www.nhlbi.nih.gov/health/sickle-cell-disease/treatment#:~:text=bone%20marrow%20transplant-,A%20blood%20and%20bone%20marrow%20transplant%20is%20currently%20the%20only,match%20to%20be%20a%20donor
vii Kanter J, Thompson AA, Pierciey FJ Jr, Hsieh M, Uchida N, Leboulch P, Schmidt M, Bonner M, Guo R, Miller A, Ribeil JA, Davidson D, Asmal M, Walters MC, Tisdale JF. Lovo-cel gene therapy for sickle cell disease: Treatment process evolution and outcomes in the initial groups of the HGB-206 study. Am J Hematol. 2023 Jan;98(1):11-22. doi: 10.1002/ajh.26741. Epub 2022 Oct 10. PMID: 36161320; PMCID: PMC10092845.
viii“bluebird bio Submits Biologics License Application (BLA) to FDA for lovotibeglogene autotemcel (lovo-cel) for Patients with Sickle Cell Disease (SCD) 12 years and Older with a History of Vaso-Occlusive Events”, Accessed 6/6/2023, https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-submits-biologics-license-application-bla-fda-0
ix Sickle Cell Disease, Accessed, 6/5/2023, https://www.vrtx.com/our-science/pipeline/sickle-cell-disease/
x de la Fuente J, Locatelli F, Frangoul H, Corbacioglu S, Wall D, Cappellini M, de Montalembert M, Kattamis A, Lobitz S, Rondelli D, Sheth S, Steinberg M, Walters M, Bobruff Y, Simard C, Song Y, Zhang L, Sharma A, Imren S, Hobbs B, Grupp S. 5612617 EFFICACY AND SAFETY OF A SINGLE DOSE OF EXAGAMGLOGENE AUTOTEMCEL FOR TRANSFUSION-DEPENDENT-THALASSEMIA AND SEVERE SICKLE CELL DISEASE. Hemasphere. 2023 Apr 10;7(Suppl ):2-3. doi: 10.1097/01.HS9.0000928144.02414.84. PMCID: PMC10112499.
xi Shapiro, L, “Vertex, CRISPR complete BLA for US approval of exa-cel”, Accessed 6/6/2023, https://sicklecellanemianews.com/news/vertex-crispr-complete-bla-exa-cel-sickle-cell-disease/